
Structural Biology
Better Models for the Crystals of Biological Macromolecules
In crystallography, the R-value reports on how well a model agrees with the experimental data. In small molecule crystallography, R-values of 3% are routinely reached. However, for biological macromolecules, R-values are usually around 24%. Something is amiss; our current models of macromolecular crystal structures are incomplete, partially incorrect, or the data have errors that we are not accounting for. These shortcomings particularly impede the determination of challenging structures, such as membrane proteins, where often only low resolution data are available. In these cases, the poor phase estimates currently obtainable result in noisy electron density maps that are difficult or even impossible to interpret.
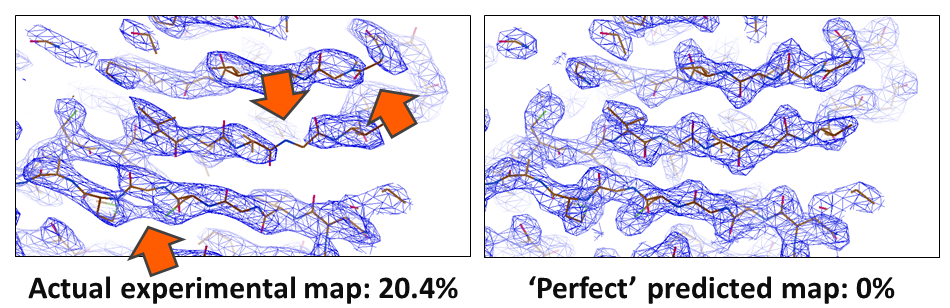
Potential causes for the large discrepancy between our atomic models and data sets are heterogeneity and atoms being treated as uncharged in Cryo-EM, solvation and protein dynamics, and perhaps lattice vibrations in crystallography. However, we need unbiased maps in order to find out: In collaboration with Armin Wagner's group at Diamond Lightsource in our current DFG project, we shed light on these problems and improve our molecular models of biological crystals, so that we cannot only solve borderline cases but also improve all known structures. (And get better training data for AI-based prediction, too!)
Back to Research